Three 2026 deadlines in EU MedTech regulation are converging on the same document

Sofija Mamuchevski
Product & Growth Lead | AI Products | Demos, Outreach, Partnerships
Sofija Mamuchevski is Product & Growth Lead at DocuGenius, where she helps organizations modernize complex document workflows through automation, operational standardization, and compliance-focused processes. With more than 10 years of experience across software development, product management, and Agile delivery, she writes about document automation, operational efficiency, product strategy, and workflow transformation in regulated and document-heavy industries.
Reading time
7 min read
Published
May 13, 2026
Updated
June 2, 2026
As a Product & Growth Lead, I keep seeing the same challenge across MedTech teams: the same documentation is repeatedly reorganized and reviewed for different regulatory processes — and that pressure is only growing.

I work as a Growth Lead at DocuGenius, so I spend a lot of time around document workflows, operational processes, and the challenges teams face when dealing with complex documentation at scale.
Over the last few months, one topic kept coming up repeatedly in conversations across MedTech regulatory workflows. Different teams, different companies — but very similar frustrations around documentation complexity, review expectations, and growing operational pressure.
That pushed me to spend more time reading through the actual regulations, implementation timelines, and source documents behind the changes happening in Europe right now.
Most discussions treat these as separate initiatives. But in practice, they all touch the same documentation: clinical evaluation reports, post-market follow-up reports, periodic safety updates, vigilance data, and real-world evidence. Same evidence. Different reviewers. Different expectations.
And honestly, that's the part I don't think enough people are talking about yet.
One thing we consistently see across document-heavy industries is that teams rarely struggle with generating information itself. The real challenge is repeatedly restructuring and adapting the same information for different workflows, reviewers, and compliance expectations.
That's really what this article is about.
1. The MDR/IVDR simplification package
It looks like the EU is finally trying to reduce some of the pressure and complexity around medical device regulations in Europe. Back in December 2025, the European Commission published a proposal to simplify parts of MDR and IVDR — meant to reduce administrative overhead, and hopefully improve timelines with Notified Bodies.
They also introduced proposed EU-wide target timelines for reviews, audits, and certificate issuance.
From the outside, that all sounds pretty positive.
A big part of this conversation is also happening right now at MedTech Forum 2026 in Stockholm. One of the key sessions this week — "Future-proofing MDR & IVDR: building a competitive and trusted EU system" — brings together European Commission representatives, Notified Body leadership, national regulatory authorities, and global MedTech manufacturers.
That's important because it shows how central this topic has become across the industry. The pressure around regulatory timelines, documentation complexity, and operational scalability is no longer just a compliance discussion — it's becoming a broader operational and competitiveness issue for MedTech companies across Europe.
But honestly, looking at this more from the operational and product side, I don't think the biggest issue has ever been just "too many documents."
What I keep hearing from teams is that the painful part is the constant rework. The same information gets rewritten, reorganized, and reformatted over and over depending on the reviewer, submission, or workflow.
So to me, the more interesting shift is not simply reducing documentation. It's moving toward processes that are more predictable and structured. If documentation can be organized once, traced properly, and reused across different workflows, that probably removes way more friction than just cutting a few requirements here and there.
2. Joint Clinical Assessments are starting for medical devices
In 2026, some medical devices will officially enter the new Joint Clinical Assessment (JCA) process in Europe for the first time.
The first group will be small and limited to certain higher-risk devices. But the important part is not how many devices are included yet — it's how the documentation will start being reviewed.
Most companies will not need completely new evidence. The same clinical evaluation reports, follow-up reports, safety updates, and supporting documentation already exist today.
What changes is the audience.
Instead of one reviewer or one national authority, multiple European assessment groups may now look at the same evidence. That means the information will need to be more structured, easier to trace, and easier to reuse across different review processes.
3. Europe is also preparing the future rules for health data reuse
At the same time, Europe is building the operational framework behind the European Health Data Space (EHDS).
A lot of people still think of EHDS mainly as a hospital or patient-data initiative. But it also affects how MedTech companies structure and manage post-market data, vigilance information, and real-world evidence over time.
In simple terms, the way documentation and data are organized today may directly affect how reusable and accessible they become in future European data-sharing and review systems.
That's why 2026 matters so much. A lot of the structure and expectations behind these future workflows are being defined right now.
Why this looks like three separate problems — but really isn't
If you look inside most MedTech regulatory workflows today, you'll usually find the same core documentation:
- Clinical evaluation reports (CER)
- Post-market clinical follow-up reports (PMCF)
- Periodic safety update reports (PSUR)
- Vigilance data
- Real-world evidence
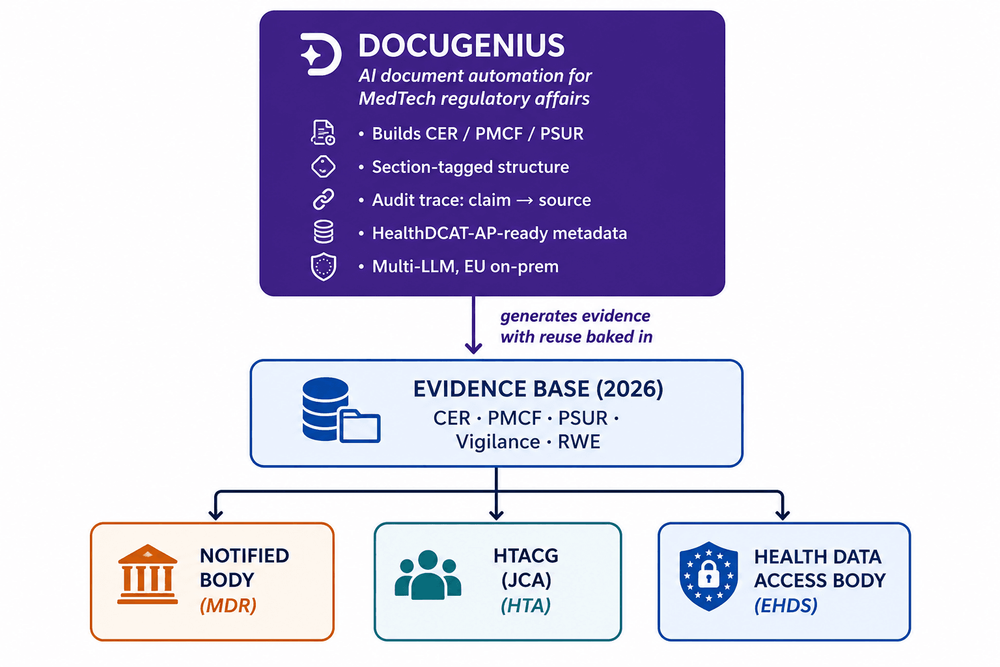
What's changing in 2026 is not necessarily the documents themselves. It's the number of different people and systems reviewing them. The same documentation may now need to support:
- Regulatory reviews by Notified Bodies under MDR/IVDR
- Joint Clinical Assessments across Europe (HTA regulation)
- Future European health data and secondary-use pathways (EHDS)
Different reviewers. Different expectations. Same evidence base.
And that's where the operational pressure starts growing — because every group wants the information structured, traceable, and reusable in slightly different ways.
The 2026 evidence flow
This shift is already becoming visible across MedTech. The challenge is no longer only about creating documentation. It's about making the same information easier to structure, trace, review, and reuse across different regulatory workflows.
Right now, many teams still spend huge amounts of time reformatting and reorganizing the same evidence for different reviewers and processes. That's why more structured and reusable workflows will likely become increasingly important over the next few years.
What I'd focus on this quarter
Just three things:
- Review existing reports and check how easily claims can be traced back to supporting evidence.
- Pay attention to the European Health Data Space discussions happening now — future expectations around reusable health data are being defined today.
- Look honestly at how reusable your current documentation really is across different review processes.
One last thing
I'm not writing this as a regulatory insider. I work on the technology side, close to the operational challenges teams deal with around complex documentation workflows.
But one thing keeps coming up in conversations across the industry: expectations around documentation quality, traceability, and reusability are evolving faster than many organizations expected.
2026 may be the year those gaps become much more visible in real review and submission workflows.
If your team is seeing similar challenges, I'd genuinely be interested to hear how you're approaching them.
Where DocuGenius fits into this
To be completely transparent — this is also the exact problem space we work in at DocuGenius.
What we kept noticing is that many teams are not actually struggling with the knowledge itself. They already have the reports, the evidence, the expertise, and the documentation.
The exhausting part is everything happening around it: reorganizing information, rebuilding the same workflows, manually reviewing long PDFs, and adapting the same content again and again for different reviewers and processes.
That's the gap we're trying to help with.
At DocuGenius, we focus on turning long, complex documentation into more structured and reusable workflows, so teams spend less time manually rebuilding the same processes from scratch.
One capability we believe will become increasingly important over the next few years is the ability to generate structured workflows directly from existing PDF documents — instead of manually translating hundreds of pages into review logic every single time.
Because honestly, most teams do not need more information. They need less friction around the information they already have.
Sources
- Commission proposal COM(2025) 1023 final — Simplification of MDR/IVDR rules
- RAPS — EU officials detail proposed MDR and IVDR revisions
- Joint Clinical Assessments — European Commission DG Health
- Regulation (EU) 2025/327 — European Health Data Space (EHDS)
- EHDS dataset descriptions consultation — Initiative 15673
- HealthDCAT-AP — European Health Information Portal
- EMA Management Board December 2025 — DARWIN EU network update
- The MedTech Forum 2026
Sofija Mamuchevski
Product & Growth Lead | AI Products | Demos, Outreach, Partnerships
Sofija Mamuchevski is Product & Growth Lead at DocuGenius, where she helps organizations modernize complex document workflows through automation, operational standardization, and compliance-focused processes. With more than 10 years of experience across software development, product management, and Agile delivery, she writes about document automation, operational efficiency, product strategy, and workflow transformation in regulated and document-heavy industries.